Terminology and Information Model¶
This is a DRAFT information model that was developed as part of the GA4GH GKS SA subgroup’s exercise to model sequence features and transcripts. It is a work in progress.
This information model was derived from the draft conceptual and logical models that were developed as foundational work for this effort. Please reference those models in addition to this documentation (which is incomplete).
Note: Some elements are sourced from the Variation Representation Specification (VRS) and others from the HL7 Clinical Genomics FHIR Molecular Sequence Resource. This draft model was aligned to those specifications as much as possible while still achieving the goals of the SA modeling exercise.
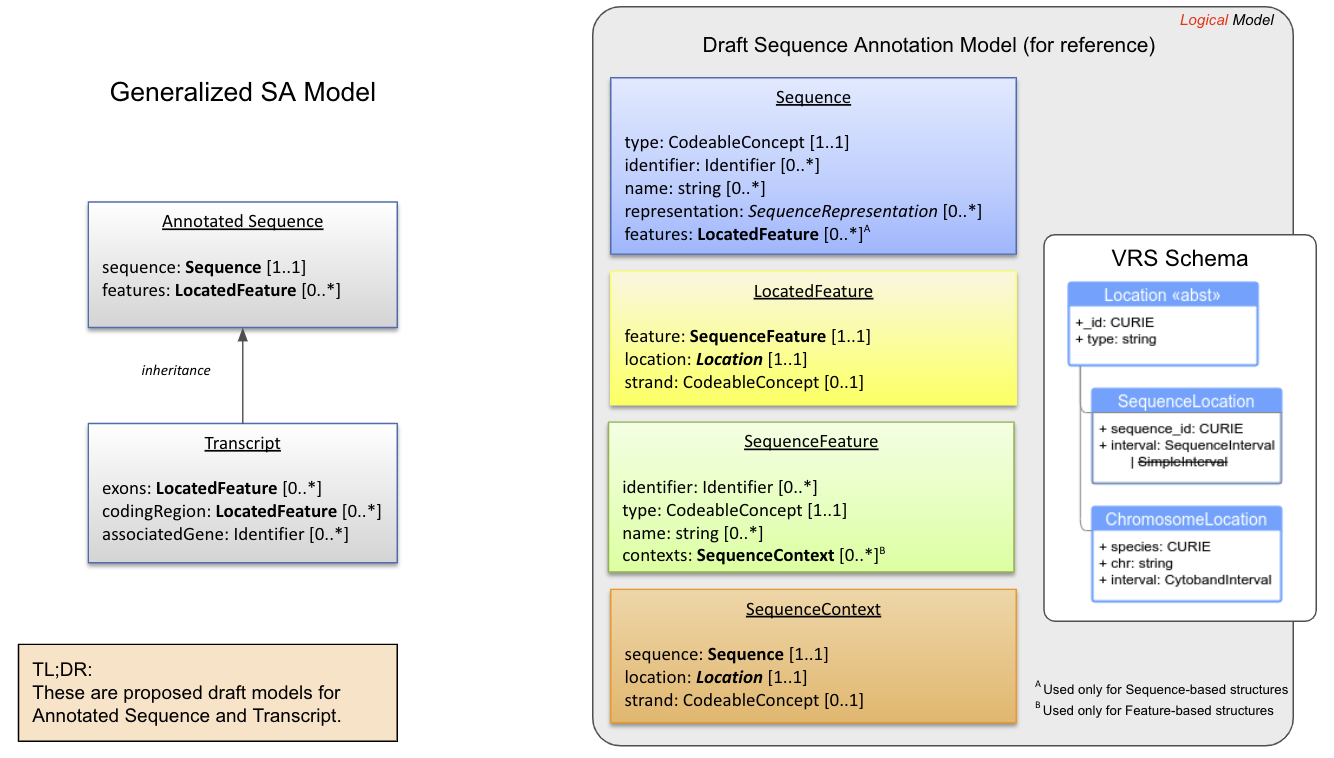
Draft SA Model¶
Legend DRAFT model of Annotated Sequence, Transcript, and building blocks (including Sequence and SequenceFeature). See the conceptual and logical models for rationale, design choices, and examples.
Core Classes¶
Sequence¶
The definition of Sequence is conceptually identical to VRS Sequence, but the information model is more detailed.
A sequence is a contiguous, linear polymer of nucleic acid or amino acid Residues. Sequences are the prevalent representation of these polymers, particularly in the domain of variant representation.
Computational Definition
A character string of Residues that represents a biological sequence using the conventional sequence order (5’-to-3’ for nucleic acid sequences, and amino-to-carboxyl for amino acid sequences). IUPAC ambiguity codes are permitted in Sequences.
Information Model
The Sequence class represents the concept of a particular sequence rather than its instantiation in a particular database or serialization. The Sequence class provides convenience attributes to capture human-readable names and identifiers that are associated with a sequence, but the representation of the sequence itself (the linear string of residues) is captured using the SequenceRepresentation abstract data type (defined by the FHIR Molecular Sequence Resource, which are similar to the VRS SequenceExpression classes).
Field |
Type |
Limits |
Description |
|---|---|---|---|
type |
[1..1] |
The type of sequence (e.g., DNA, RNA, protein). The value of this attribute should represent a term in an ontology (e.g., Sequence Ontology). |
|
identifier |
[0..*] |
Identifier(s) for the sequence. Identifiers are used for cross-referencing sequence concepts only, not for capturing the sequence itself (which is done with the representation attribute). Note the complex datatype includes attributes to capture both the identifer and the system or namespace that assigned the identifier. Identifiers MUST be unique within a system or namespace. |
|
name |
string |
[0..*] |
Name(s) for the sequence. Names are intended to be for human-readable purposes only and are not guaranteed to uniquely specify a sequence. |
representation |
SequenceRepresentation |
[0..*] |
Representation(s) of the sequence. All representations for a given sequence MUST resolve to the same literal string. |
features |
[0..*] |
A list of annotated features on the sequence. This attribute should be used only by Sequence-based structures. |
LocatedFeature¶
A located feature is a mapping of a sequence feature to a location on a sequence.
Computational Definition
TBD
Information Model
A mapping between a given sequence feature and its location on a given sequence.
Field |
Type |
Limits |
Description |
|---|---|---|---|
feature |
[1..1] |
The feature at the given location |
|
location |
[1..1] |
The location of the feature |
|
strand |
[0..1] |
An indicator specifying whether the feature is on the forward or reverse strand of a double-stranded sequence. If not set, the feature is assumed to be on the forward strand (by convention). If the sequence is single-stranded and this attribute is set, it must not be set to “reverse”. |
SequenceFeature¶
A sequence feature is a structural or functional feature that can be annotated on a Sequence.
Computational Definition
TBD
Information Model
A structural or functional feature that can be annotated on a Sequence at a defined location (interval). A given feature is not unique to a single Sequence and can be mapped to different locations on different Sequences through contextualization.
- Examples of Sequence Features include:
Gene locus
Exon
Intron
5’ or 3’ UTR
Transcription start site
Translation start or stop sites
Coding sequence (CDS)
Codon
Transcription factor binding site
Post-translational modification site
Splice donor/acceptor site
PolyA site
Sequence Features are first-class entities that can stand on their own as independent entries in knowledge bases and be used as the subject of VA statements.
Field |
Type |
Limits |
Description |
|---|---|---|---|
identifier |
[0..*] |
Identifier(s) for the sequence feature. Note the complex datatype includes attributes to capture both the identifer and the system or namespace that assigned the identifier. Identifiers MUST be unique within a system or namespace. |
|
name |
string |
[0..*] |
Name(s) for the sequence feature. Names are intended to be for human-readable purposes only and are not guaranteed to uniquely specify a sequence feature. |
type |
[1..1] |
The type of feature. The value of this attribute should refer to a term in an ontology (e.g., Sequence Ontology). |
|
contexts |
[0..*] |
A list of sequence contexts for the feature. This attribute should be used only by Feature-based structures. |
SequenceContext¶
A sequence context is the definition of a location on a sequence.
Computational Definition
TBD
Information Model
The definition of a location on a given sequence. This mapping provides sequence context (contextualization) for a parent entity (e.g., a feature).
Field |
Type |
Limits |
Description |
|---|---|---|---|
sequence |
[1..1] |
The Sequence that provides the context for the mapped location. |
|
location |
[1..1] |
A location that defines a region (interval) on the given Sequence. |
|
strand |
[0..1] |
An indicator specifying whether the feature is on the forward or reverse strand of a double-stranded sequence. If not set, the feature is assumed to be on the forward strand (by convention). If the sequence is single-stranded and this attribute is set, it must not be set to “reverse”. |
Annotated Sequence¶
An annotated sequence is a sequence that contains annotations (e.g., features).
Annotated Sequence¶
Computational Definition
TBD
Information Model
Conceptually, an annotated sequence is a sequence with associated annotations (e.g., features). Structurally, the class is a container for a Sequence and located features (and because of this an Annotated Sequence is a complex object that is neither a Sequence nor a Feature). This class may serve as a generalized parent class that can be specialized to support complex types of annotated sequences (e.g., transcripts).
Field |
Type |
Limits |
Description |
|---|---|---|---|
sequence |
[1..1] |
The Sequence that is annotated with features. |
|
features |
[0..*] |
A list of annotated features on the Sequence. |
Transcript¶
A transcript is a type of annotated sequence.
Computational Definition
TBD
Information Model
A transcript is a type of annotated sequence. It represents a single-stranded RNA sequence that may contain structural features (e.g., exons) and functional features (e.g., coding region). Because the concept of transcript is so widely used, and because the concepts of sequence, exon, and gene are so closely intertwined with it, an explicit model for Transcript was developed.
Field |
Type |
Limits |
Description |
|---|---|---|---|
exons |
[0..*] |
A list of sequence features representing the exons of the transcript. |
|
codingRegion |
[0..*] |
A list of sequence features representing the coding region of the transcript. |
|
associatedGenes |
identifier |
[0..*] |
A list of gene(s) associated with the transcript. Note: This structure provides support for fusions, prokaryotic operons, etc, but it does not specify which region(s) or feature(s) within the transcript are associated with a particular gene. |